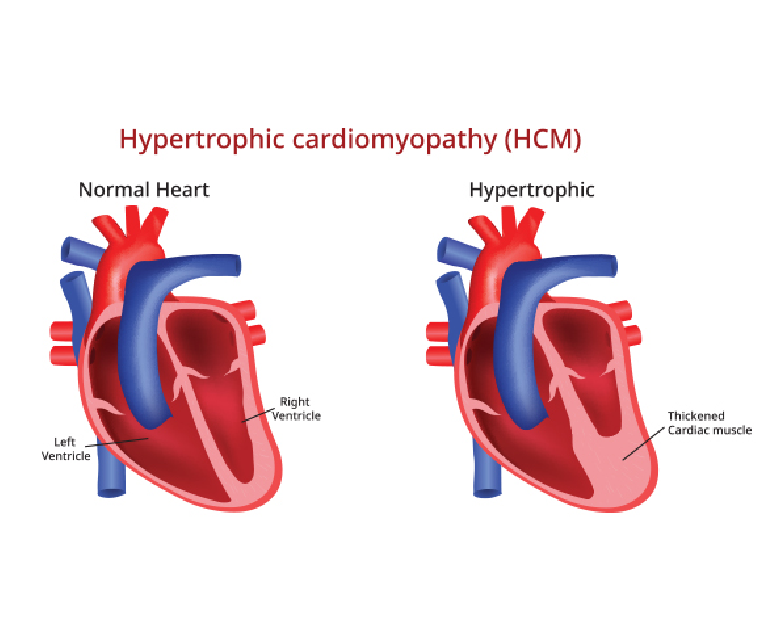
Hypertrophic cardiomyopathy (HOCM) is a complex and potentially life-threatening heart condition characterized by abnormal thickening (hypertrophy) of the heart muscle. It is one of the most common genetic heart disorders, affecting people of all ages and backgrounds.
Causes
Hypertrophic cardiomyopathy is primarily caused by mutations in genes responsible for the structure and function of the heart muscle. These genetic mutations lead to abnormal growth and arrangement of the cardiac muscle cells, resulting in the characteristic thickening of the heart muscle.
HOCM is usually inherited in an autosomal dominant pattern, meaning that a child has a 50% chance of inheriting the condition if one of the parents is affected. However, spontaneous gene mutations can also occur, leading to the development of HOCM in individuals without a family history of the condition.
Symptoms
The symptoms of hypertrophic cardiomyopathy can vary widely among individuals. Some may remain asymptomatic throughout their lives, while others may experience severe symptoms. Common symptoms include shortness of breath, chest pain, heart palpitations, fatigue, dizziness, fainting, and lightheadedness. These symptoms can worsen during physical exertion or strenuous activities and may be indicative of an increased risk of sudden cardiac arrest, especially in young athletes.
Diagnosis
The diagnosis of hypertrophic cardiomyopathy involves a comprehensive evaluation of the patient’s medical history, a physical examination, and a variety of diagnostic tests. These tests may include electrocardiography (ECG), echocardiography, cardiac MRI, exercise stress tests, and genetic testing. ECG can help identify abnormal electrical patterns in the heart, while echocardiography and cardiac MRI provide detailed images of the heart’s structure and function. Genetic testing is performed to identify specific gene mutations associated with HOCM.
Treatment
The management of hypertrophic cardiomyopathy focuses on relieving symptoms, preventing complications, and reducing the risk of sudden cardiac arrest. Treatment plans are tailored to individual patients based on their symptoms, risk factors, and disease progression. Medications such as beta-blockers, calcium channel blockers, and anti-arrhythmic drugs are commonly prescribed to control heart rate, rhythm, and blood pressure.
In some cases, surgical procedures like septal myectomy or alcohol septal ablation may be recommended to relieve obstruction of blood flow from the heart. Implantable cardioverter-defibrillators (ICDs) may also be used in high-risk patients to prevent sudden cardiac arrest.
Prognosis
The prognosis for individuals with hypertrophic cardiomyopathy varies depending on the severity of the disease, the presence of complications, and the effectiveness of treatment. Many people with HOCM lead normal lives with minimal symptoms and a low risk of complications. Regular monitoring, adherence to treatment plans, and lifestyle modifications, such as avoiding strenuous activities, can help improve outcomes and reduce the risk of complications.
Conclusion
Hypertrophic cardiomyopathy is a complex heart condition characterized by abnormal thickening of the heart muscle. It is primarily caused by genetic mutations and can present with a range of symptoms and complications. Early diagnosis, thorough evaluation, and personalized treatment plans are essential for managing HOCM effectively. With appropriate medical care and lifestyle modifications, individuals with hypertrophic cardiomyopathy can lead fulfilling lives while minimizing the risks associated with the condition.