

News
Our News Section offers timely articles, breaking news, and expert insights on medical advancements, new treatments, and healthcare policies.

Lifestyle Changes after CABG
Coronary Artery Bypass Grafting (CABG) surgery is often part of comprehensive treatment for coronary artery disease. After undergoing CABG, individuals…

Psychological Impact
The psychological impact of undergoing Coronary Artery Bypass Grafting (CABG) surgery can vary from person to person. Factors such as…
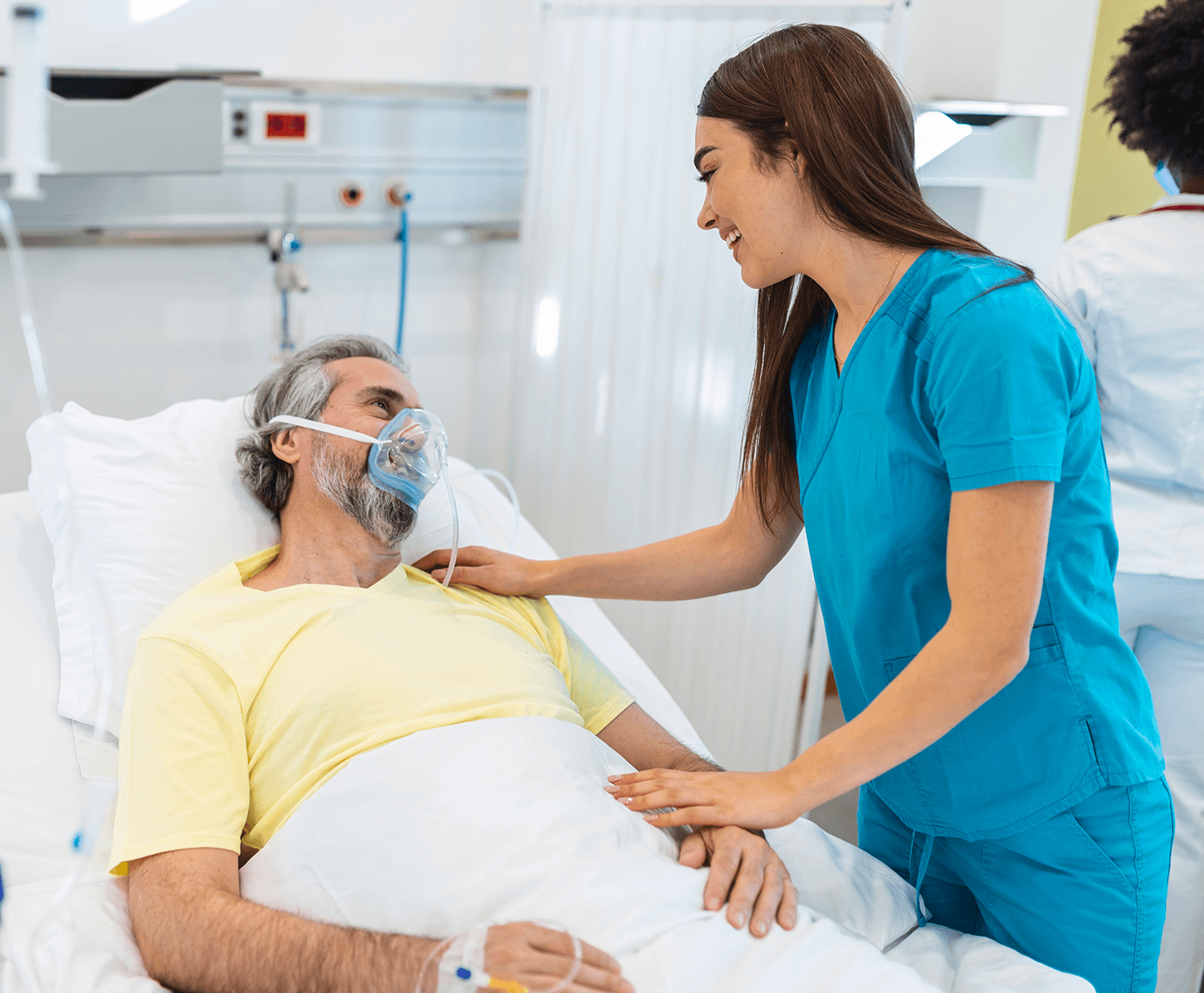
Preoperative and Postoperative Care
The preoperative and postoperative care for Coronary Artery Bypass Grafting (CABG) is crucial for the overall success of the procedure…
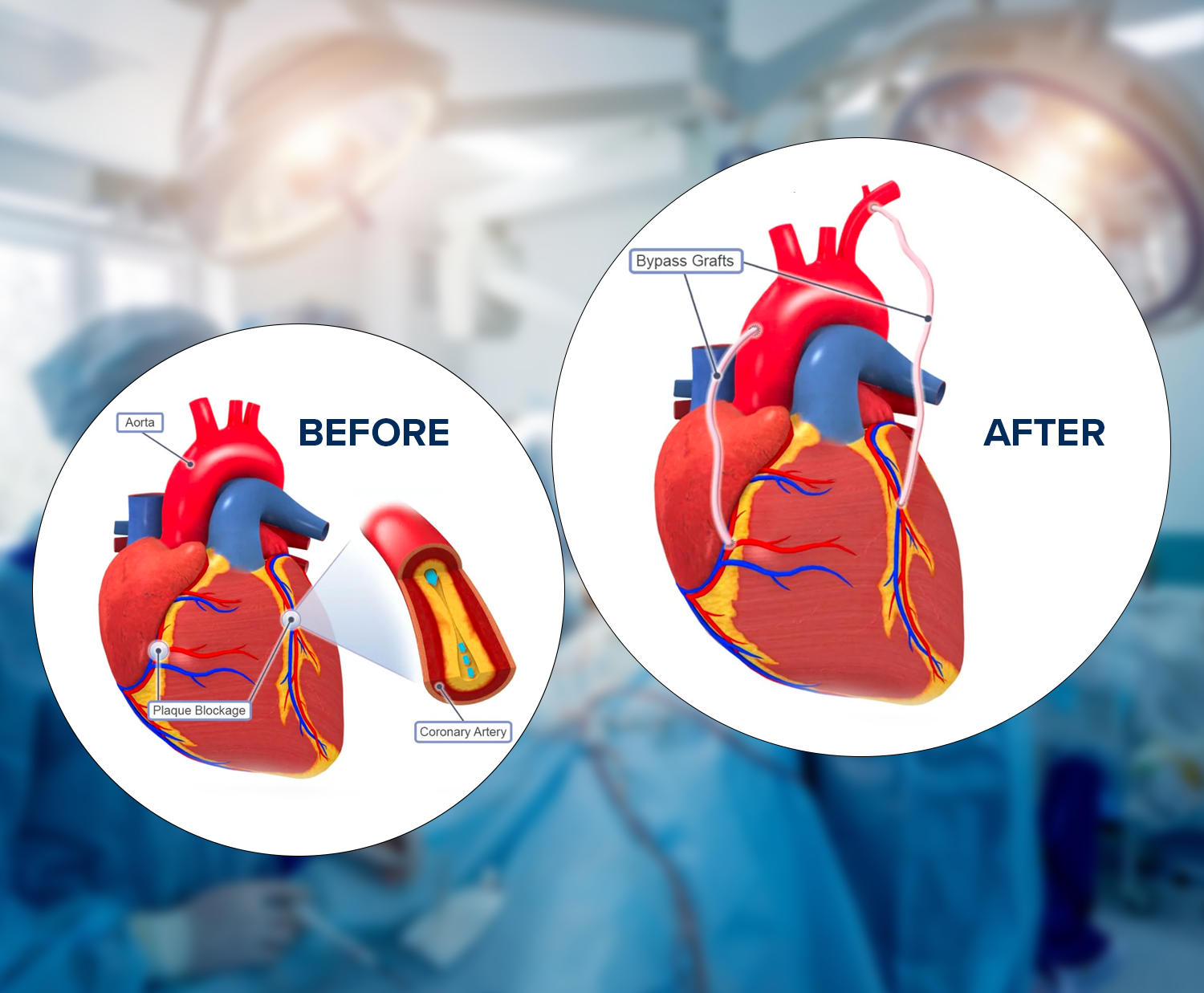
CABG Procedure Overview
Coronary Artery Bypass Grafting (CABG) is a surgical procedure commonly performed to treat coronary artery disease (CAD), a condition in…
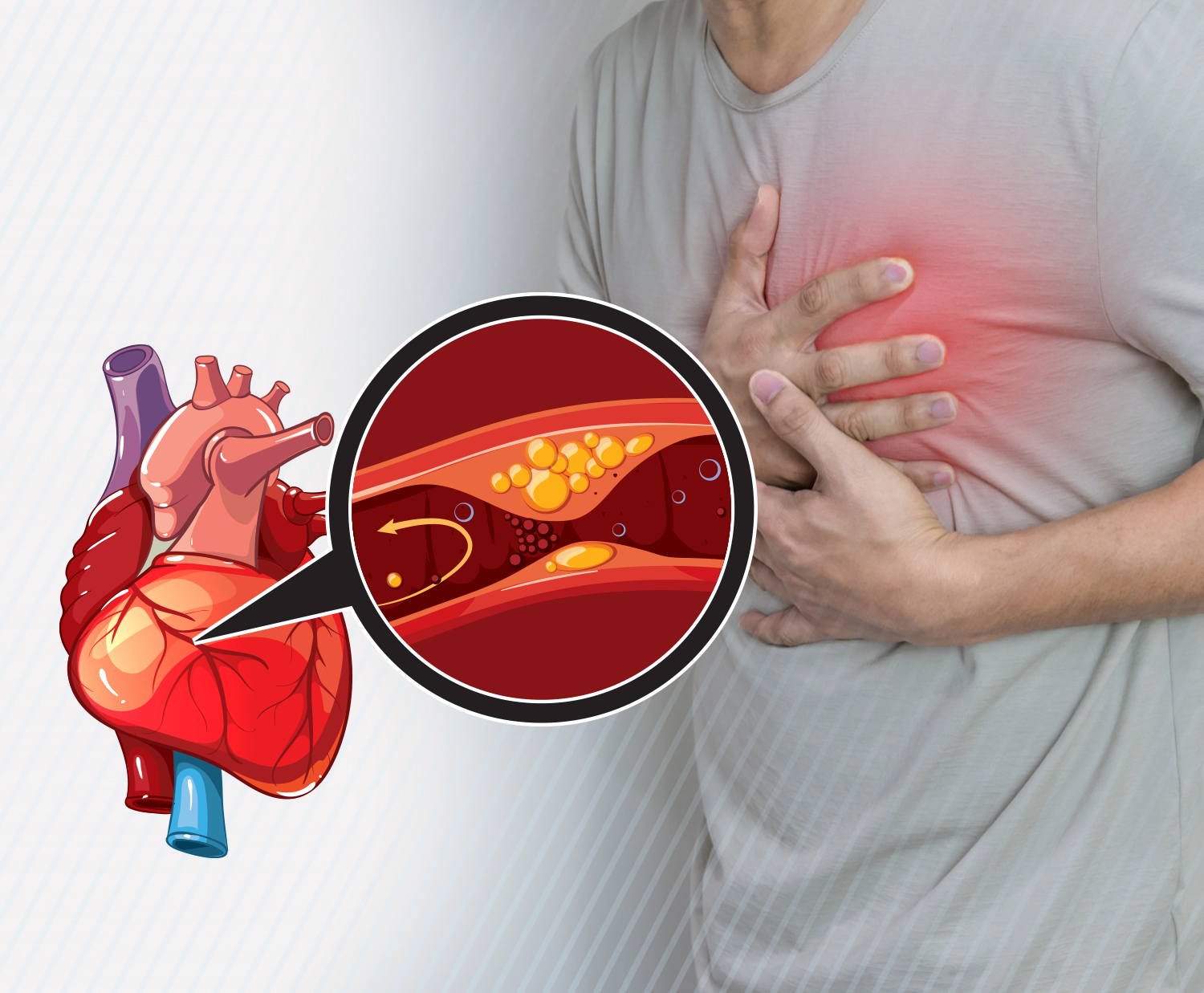
Indications for CABG
Coronary Artery Bypass Grafting (CABG) is a surgical procedure used to treat coronary artery disease (CAD), a condition where the…

How Heart Disease and Autoimmune Disorders related
Heart disease and autoimmune disorders can be related through various mechanisms, and the relationship between them is complex. Here are…
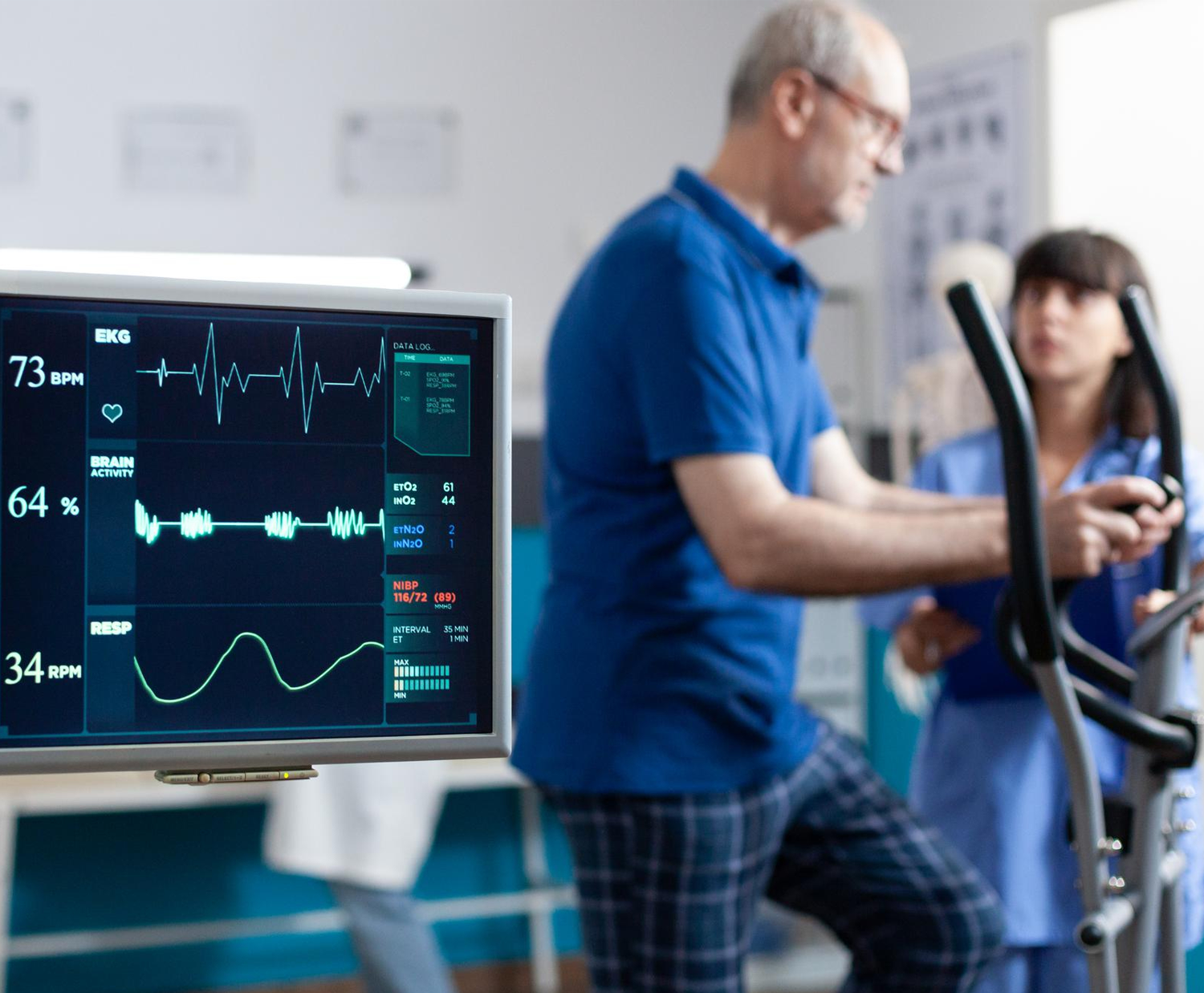
What is Cardiac Rehabilitation
Cardiac rehabilitation (CR) is a structured and medically supervised program designed to help individuals recover from various heart-related conditions and…
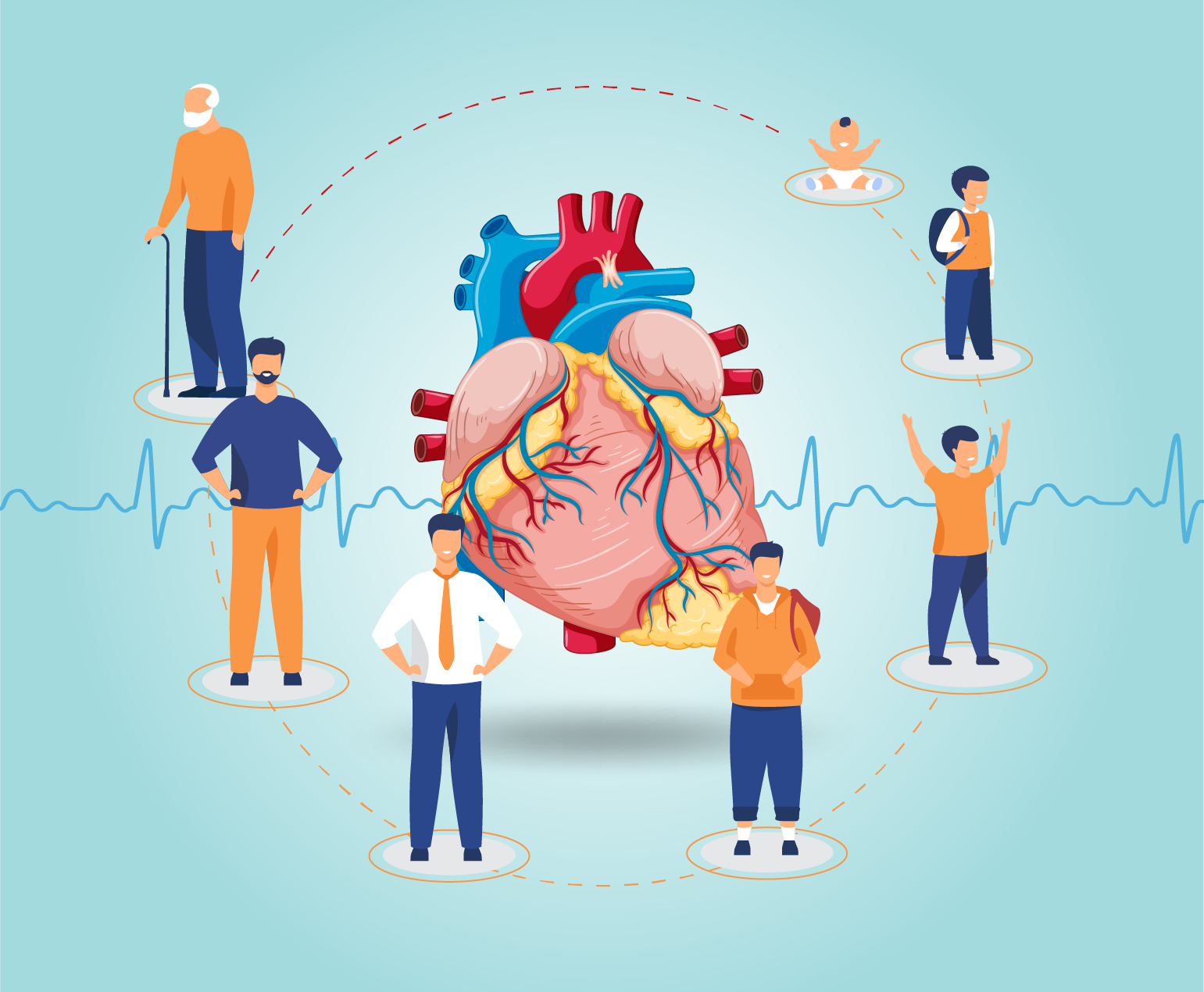
Cardiovascular Health in Different Age Groups
Cardiovascular health is important at every stage of life, and different age groups have specific considerations for maintaining a healthy…

Genetics and Heart Disease
Genetics and heart disease are intricately connected, and the field of study that explores this relationship is known as cardiovascular…
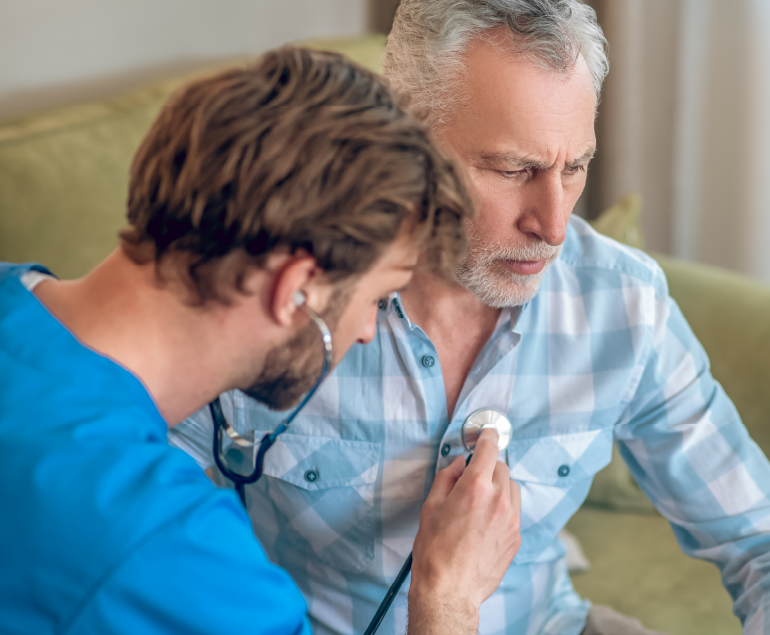
Aging and Heart Disease
Aging and Heart Disease are interconnected in several ways. As individuals age, their chances of developing heart disease increase. Aging…
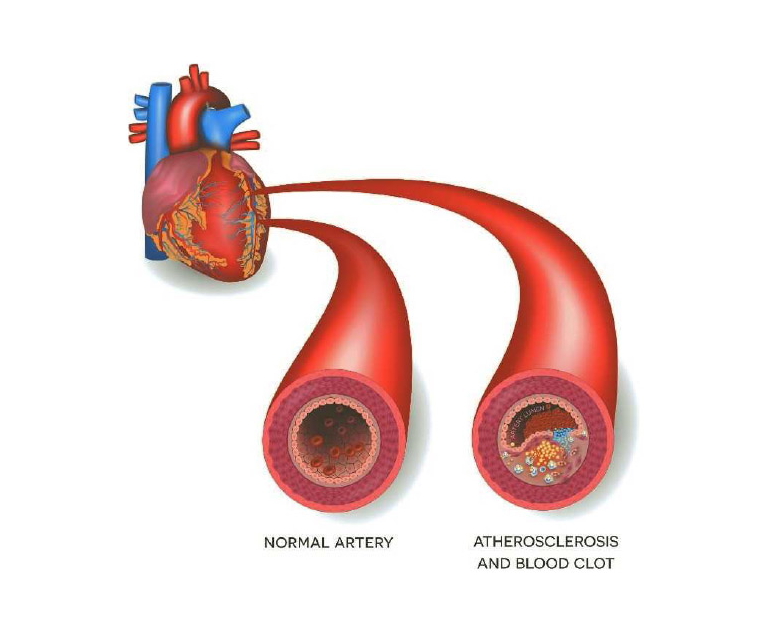
Atherosclerosis
Atherosclerosis is a progressive condition in which the arteries become narrowed and hardened due to the buildup of plaque on…
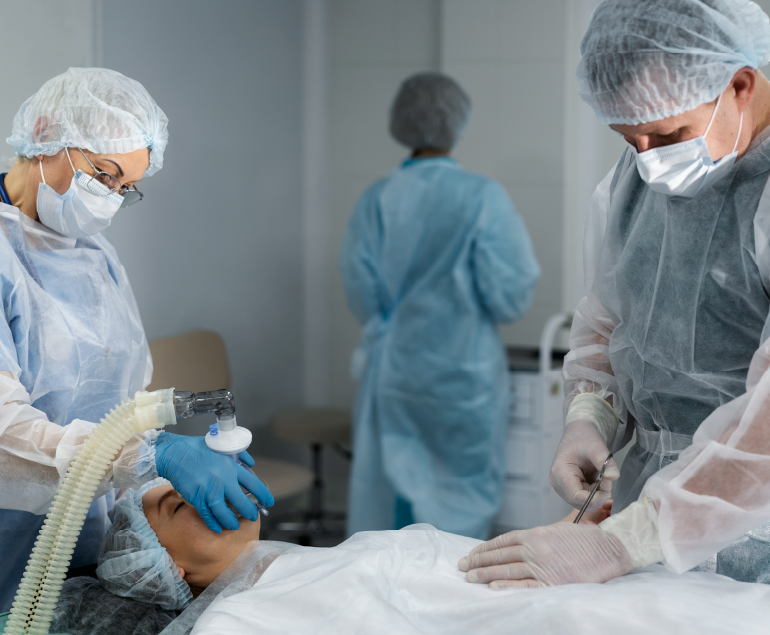
Heart Transplantation Procedure
A heart transplant is a complex surgical procedure that involves replacing a diseased or failing heart with a healthy donor…

Heart Health Precautions during the Winter
Maintaining heart health during the winter is important, as colder temperatures can affect the cardiovascular system. Here are some precautions…

Air Pollution & Heart Disease
Air pollution is considered to be a silent killer that affects our daily lives. From the exhaust fumes of vehicles…

Pediatric Heart Infections
Pediatric heart infections, although relatively rare, can have serious and potentially life-threatening consequences for children. These infections affect the heart…