

Cardiac Amyloidosis
Cardiac amyloidosis is a rare and potentially fatal disorder that arises from the abnormal accumulation of amyloid protein in the heart tissues, leading to compromised cardiac function.
Nature of Cardiac Amyloidosis
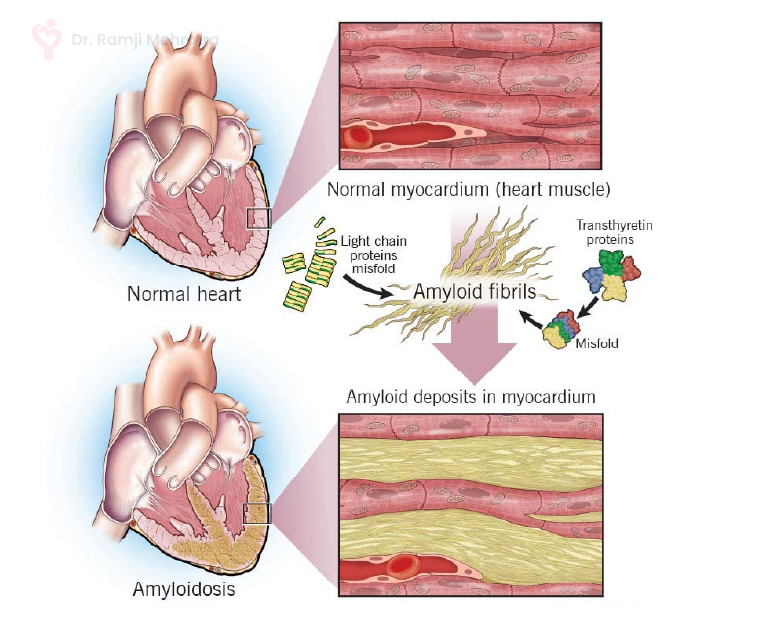
Cardiac amyloidosis stems from the buildup of amyloid fibrils, which are insoluble protein aggregates that accumulate in various organs, including the heart. These amyloid deposits infiltrate the heart muscles, causing them to become stiff, thickened, and less compliant. Over time, this impairs the heart’s ability to pump blood effectively, leading to heart failure and other cardiovascular complications.
Subtypes of Cardiac Amyloidosis
There are different subtypes of cardiac amyloidosis, each originating from distinct precursor proteins. The two most prevalent subtypes are:
- AL Amyloidosis (Immunoglobulin Light Chain Amyloidosis): This form arises when abnormal plasma cells in the bone marrow produce excessive amounts of light chains, a component of antibodies. These light chains then accumulate as amyloid fibrils in various organs, including the heart.
- ATTR Amyloidosis (Transthyretin Amyloidosis): In ATTR amyloidosis, the precursor protein is transthyretin, a protein responsible for transporting thyroid hormone and retinol. Mutations or aging-related changes in transthyretin can lead to the formation of amyloid fibrils that deposit in the heart.
Challenges in Diagnosis
Cardiac amyloidosis remains a diagnostic challenge due to its overlapping symptoms with other heart conditions and its rarity. Common symptoms include fatigue, shortness of breath, swelling in the legs, and irregular heartbeats. These nonspecific signs often delay accurate diagnosis, which is further complicated by the fact that cardiac amyloidosis can coexist with other heart diseases.
Advancements in Detection
Recent advancements in imaging techniques have greatly improved the detection of cardiac amyloidosis. Cardiac MRI, nuclear imaging with bone tracers, and echocardiography can provide insights into the presence and extent of amyloid deposits in the heart. Additionally, advances in genetic testing help in identifying ATTR amyloidosis, enabling early intervention for individuals with genetic predisposition.
Management and Treatment
Managing cardiac amyloidosis involves addressing both the underlying cause and the associated symptoms. In AL amyloidosis, treatments focus on suppressing the abnormal plasma cells responsible for producing the amyloidogenic light chains. Chemotherapy and stem cell transplantation are commonly used in the treatment. For ATTR amyloidosis, treatment options include stabilizing the transthyretin protein or, in cases of hereditary ATTR, gene-silencing therapies.
